고정 헤더 영역
상세 컨텐츠
본문
작성자: 14기 김혜림
[참고자료]
https://www.youtube.com/watch?v=1YHsSFWn5OA&t=4319s
https://www.youtube.com/watch?v=MHb18hhKQsY
[이미지 출처]
[단백질 구조에 대한 사전 지식]
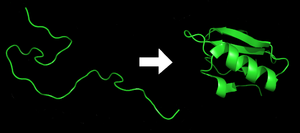
잔기 => 아미노산 => 단백질 => folding된 단백질
인간의 몸은 단백질 블럭을 쌓은 형태로 이루어짐
단백질의 기본 구조는 선
다만 이때 일직선으로 존재하는 것이 아니라, 어떠한 일정한 형태로 구겨져서(folding) 존재함
이 folding 방식에 따라 단백질 기능 방식이 정해짐
단백질은 또 다시 아미노산으로 구성됨
=> 단백질은 아미노산이 꿰어진 실
아미노산은 아미노산 잔기(residue)에 의해 그 종류가 결정됨
(* 아미노산은 공통적인 구조를 가짐: 가운데의 탄소와 질소원자를 주축으로 다른 분자들이 연결된 형태
이때 중간의 탄소 원자에 어떤 분자가 결합되었는지에 따라 20가지의 아미노산 중 하나로 결정됨)
이러한 단백질의 구조는 MRI를 통해 3차원 구조로 재구성할 수 있는데, 이 자료를 데이터베이스화 해놓은 것이 PDB(단백질 정보 은행)
하지만 모든 단백질을 실험을 통해 데이터베이스화 해놓을 수는 없음 => protein structure prediction을 통해서 해결함
: 아미노산의 전자기적 특성에 의해서 폴딩되므로 이를 이용(단백질의 아미노산 시퀀스 => 단백질의 최종 폴딩 구조)
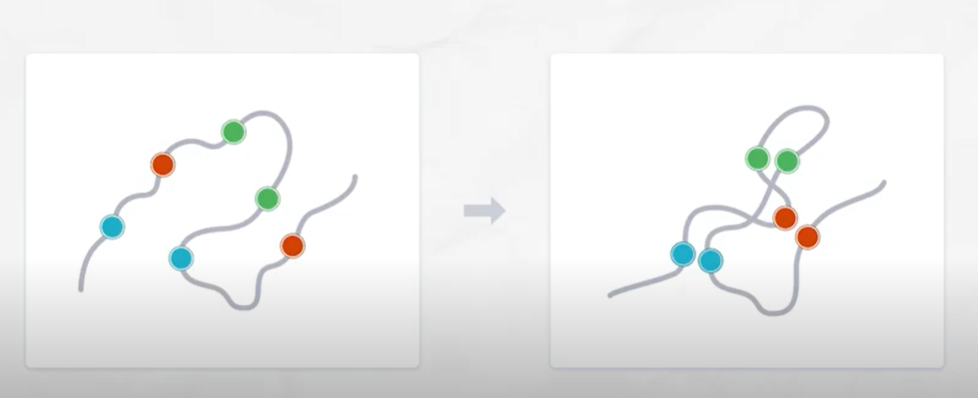
어떤 아미노산끼리 결합하는지 안다면 이들을 미리 붙여놓고 나머지 구조를 생성함으로써 훨씬 예측이 쉬워질 것
그러면 이런 접점들을 어떻게 찾느냐?

접점에서 하나의 아미노산 변형이 일어났다면 다른 하나도 마찬가지로 변화할 것 즉, 쌍으로 변화함
이를 공진화 co-evolution이라고 함
이 공진화가 이루어진 부분을 찾음으로써 단백질의 접점을 찾을 수 있음
이 진화과정을 추적하기 위해서는 유전자 정보를 정렬해야 함 : 다중서열정렬(multiple sequence alignment, MSA)

그래서 기존에는 이 MSA를 이용하여 공진화된 부분을 찾아내면 그게 접점일 가능성이 높으므로 이를 이용해서 폴딩 구조를 예측하는 방법
[MIT 강의]
(1) Methods for Protein structure prediction
| (1) Template-based modeling 기존에 자주 사용되던 방법으로, PDB에 존재하는 단백질 구조를 이용해서 단백질 구조를 예측한다. 그러나 membrane protein과 같이 PDB에 많이 등록되지 않은 단백질 구조를 예측하기에는 용이하지 않다 |
(2) Template-free modeling 최근에 사용하는 방법으로, PDB에 있는 template를 이용하지 않는다. 매우 좋은 template이 있지 않는 이상 이 방법을 주로 이용한다. |
- Old method : fragment assembly

오래된 접근법으로 fragment assembly가 있다
우선 단백질 전체 sequence를 부분(segment)으로 쪼갠다 이 하나의 seg에는 대략 9~11 개의 residue가 존재한다고 한다 이후 이 segment와 유사한 구조를 PDB에서 찾는다. 이 부분으로 쪼개는 sampling을 여러번 실시해서 전체 단백질 구조를 예측한다.
그러나 구조가 작은 단백질을 예측할 때도 많은 sampling을 해야하기 때문에 computino cost가 매우 크다고 한다.
이건 강의 외의 내용인데, 이 ppt에 보이는 구조는 정확하게 강사가 설명한 방법을 그림으로 나타낸 것은 아닌 것 같다. 대신 ROSETTA 라는 프로토콜인 것 같은데, 관련해서 찾아보다가 재밌는 기사가 있길래 각주에 달아놓겠다.1
대충 내용을 요약하자면 로제타 프로그램을 이용하여 알파폴드를 흉내낸 로제타폴드에 대한 이야기인데, 알파폴드보다 quantitive 정확성은 밀리지만 그 qualititive 정확성은 밀리지 않는다는 내용이다. 그리고 깃허브에도 공개를 해놨다고 한다. 듣기로 알파폴드2는 구조는 정확하게 안 알려졌다고 그랬는데..
- Relatively New Strategy
비교적 최신 모델으로 fragment assembly와는 사뭇 다른 접근 방법이다.
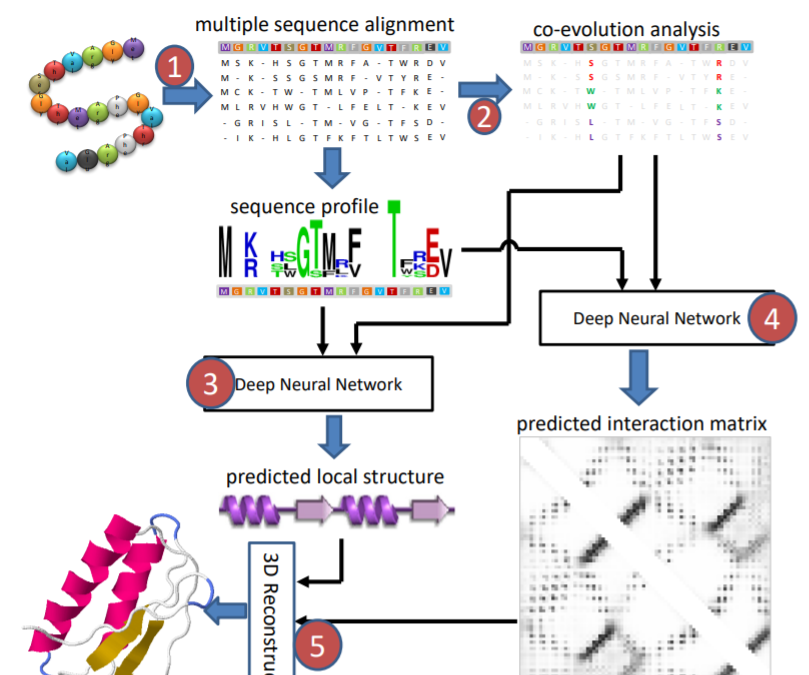
앞에서 언급했던 co-evolution 된 지점을 찾아서 단백질 구조를 예측하는 방법이다. co-evolution analysis를 통해서 local 한 정보를 predict하고(그림상 3번) 또, interaction matirx(그림상 4번)를 예측한다. interaction matrix는 residue 사이의 거리와 같은 것들에 대한 정보를 담고 있다. 여기서 중요한 건 interaction matrix를 구성하는 방법이다.
이는 contact matrix를 이용해서 구성할 수 있는데, 노드 사이의 거리가 일정 threshold를 넘기면 1, 그렇지 않으면 0 으로 처리하는 matrix라고 생각하면 쉬울 것 같다. 그래서 만약 contact matrix에서 (i,j)의 component가 1은 i와 j residue 사이의 거리는 가까움을 의미한다. 그런데 사실 요즘은 또 computing cost나 이런 걸 많이 줄일 수 있어서 contact matrix처럼 간소화한 matrix 보다는 distance matrix를 그냥 이용한다고도 한다
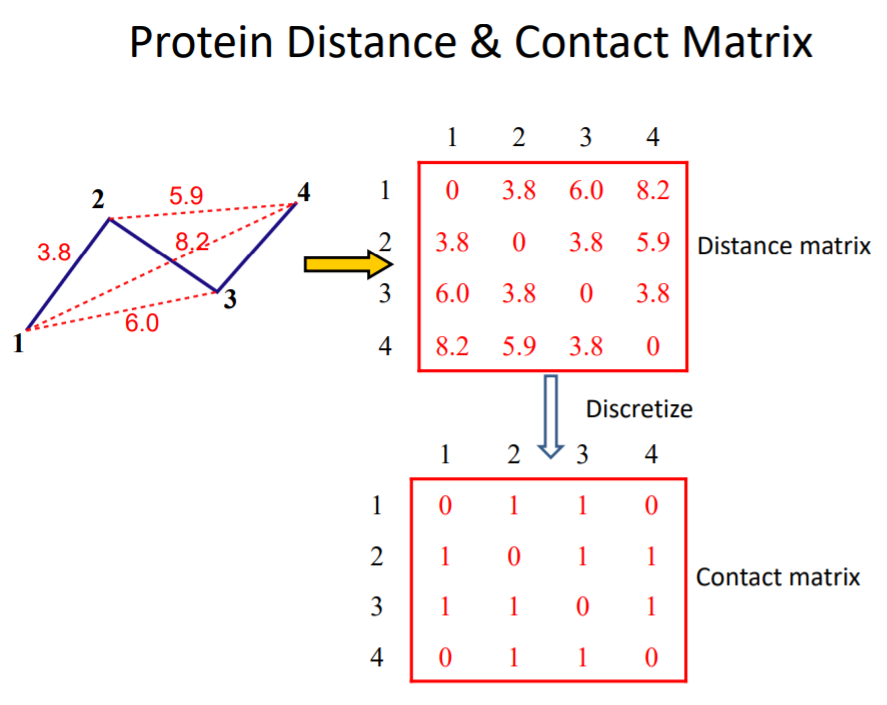
그러면 이 contact matrix/distance matrix를 어떻게 예측할 것인가??
그 전에 잠깐 다시 지금 맥락을 설명하자면 앞에서 공진화에 대해서 살펴보았다. 공진화가 일어나면 두 protein이 correlated 된다고 한다. (사실 sequence인데 정확히 이게 어딜 봐서 correlated 된 걸 알 수 있는지는 이해를 못했다) 그래서 correlation을 계산해서 어쩌구저쩌구 단백질 구조를 예측한다.
그런 어쩌구저쩌구 중에 하나가 이제
- Statistical Approach(:supervised)
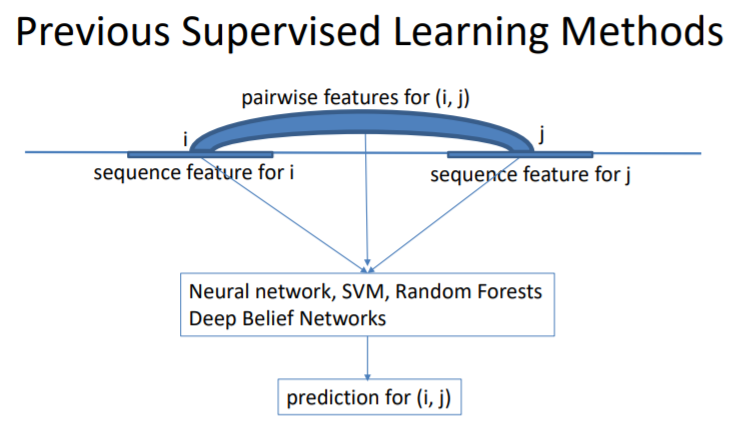
GNN에 비유하자면 node인 residue, node feature matrix인 residue feature matrix, 그리고 둘 사이를 연결하는 edge의 feature 인 mutual information인 셈이다. input으로 이것들을 이용하고 머신러닝 기법을 적용시켜서 둘 사이의 correlation 을 계산한다는 것이다. 이때의 문제점은 오직 두 개의 residue 정보만을 연산에 이용하기 때문에 다른 residue 에 대한 정보를 이용하지 못한다는 것이다. 당연히 정확도가 떨어질 수 밖에 없다.
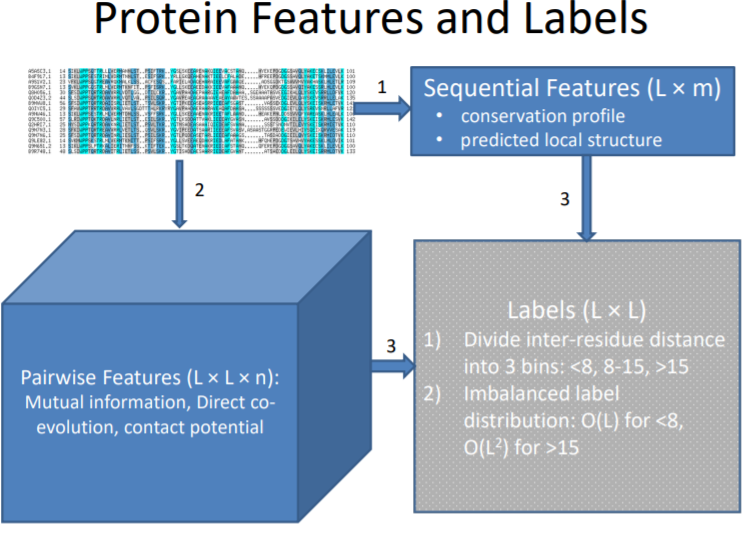
- Deep learning approach

통계적 접근에서는 서로 다른 두 residue와 관련 정보를 단순히 model 의 input으로 썼다면
이 딥러닝적 접근에서는 이 pair를 하나의 pixel값으로 취급한다.
=> 이를 통해 atomic interaction map을 하나의 그림으로 재구성한다!!
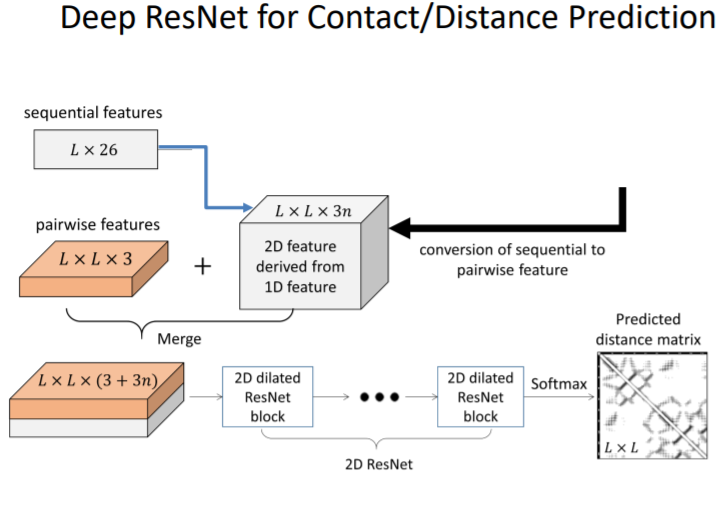
이후 ResNet을 이용해서 우리가 목표했던 대로 distance 예측을 한다.

노 -> 초 -> 빨 -> 파
: 고전적인 기법 -> 고전적 기법 + 통계적 기법 -> 고전적 기법 + 딥러닝 -> 순수 딥러닝
위까지가 Figure 6에서 설명한 (4) 이다. distance를 예측했으니 이제는 본격적으로 3d로 단백질을 재구성해야 한다.
-Builiding a 3d structure from pairwise distance
distance gemoetry방식, energy minimization이 있는데 전자는 정확도가 나자서 이용 안 한다고 한다. 그래서 후자에 대해서만 살펴보도록 하자.
일단 지금까지 우리가 살펴본 것은 2d에서 잔기들 사이의 거리이다. 이것을 3d에 표현하기 위해서는 어떻게 하는가? 쉽게 생각해서 하나의 residue를 삼차원 벡터로 표현하면 된다. 그래서 각각의 residue의 삼차원 상 좌표값을 찾기 위한 학습을 이 단계에서는 거친다고 생각하면 쉬울 것 같다.
지금까지 우리가 계산해낸 distance matrix를 이용해서 potential 함수를 계산한다. 쉽게 생각해서 하나의 loss function이라고 보면 될 것 같다. 그리고 이 potential을 최소화하는 방식으로 3d로 단백질 구조를 생성한다.
- Convert predicted distance probability to statistical potential
- Minimize potential by conformation sampling and/or gradient descent
(* potential =-log P(predicted distance)/background probability of distance)
(* 알파폴드에서는 3가지의 potential function을 이용하고 있다
residue 사이의 거리,
residue 사이의 비틀림 각도,
그리고 실제로 3d 에 construct 했을 때 부딪히는 steric clash를 피하기 위한 물리적 제약
을 반영할 수 있도록 설계한 후에
이를 합쳐 하나의 potential 함수로써 이용한다
)
- https://m.dongascience.com/news.php?idx=48847 [본문으로]
'심화 스터디 > 의료 데이터 스터디' 카테고리의 다른 글
| GNN (graph neural network) (0) | 2021.11.11 |
|---|---|
| [의료 데이터 스터디] EDRS (0) | 2021.09.30 |
| [의료 데이터 스터디] COVID-SDNet (0) | 2021.09.16 |
| 의료 데이터 스터디 소개 (0) | 2021.08.27 |



댓글 영역